üldsõnalisus
Prader-Willi sündroom on haruldane geneetiline haigus, mis põhjustab füüsilisi, käitumuslikke ja intellektuaalseid kõrvalekaldeid. Kõige iseloomulikumad kliinilised tunnused on rasvumine (ja sellega seotud haigused) ja vähenenud lihastoonus.
Füüsiline läbivaatus on tavaliselt piisav õige diagnoosi kindlakstegemiseks, kuid ka usaldusväärseid geneetilisi teste saab teha.
Kahjuks puudub veel lõplik ravi; siiski võivad mõned farmakoloogilised ja käitumuslikud vastumeetmed sellega seotud sümptomaatikat piirata.
Geneetika lühike meeldetuletus
Enne Praderi-Willi sündroomi kirjeldamist on hea teha lühike viide geneetikale.
CHROMOSOMID JA DNA
Igal tervisliku inimese rakul on 23 paari homoloogseid kromosoome : 23 on ema, see on päritud emalt ja 23 on isad või päritud isalt. Nende kromosoomide paar on seksuaalne, st see määrab inimese soo; ülejäänud 22 paari koosnevad autosomaalsetest kromosoomidest . Täielikult sisaldavad 46 inimese kromosoomi kogu geneetilist materjali, mis on paremini tuntud kui DNA . Inimese DNA-s on kirjutatud tema somaatilised tunnused, tema eelsoodumused, füüsilised võimed jne.
GENESID JA DNA MUTATSIOONID
DNA on järjestatud paljudes järjestustes, enam-vähem pikka aega, mida nimetatakse geenideks .

Joonis: geeni korraldus homoloogsete kromosoomide paari sees. Homoloogsete kromosoomide paar sisaldab spetsiifilisi geene, millel kõigil on kaks varianti, alleelid, mis omavad sama kromosomaalset positsiooni ja teevad samu funktsioone (välja arvatud mutatsioonid). Vasakul kromosoomipaaril on kaks võrdset alleeli (mõlemad sinised); paremal paaril on kaks erinevat alleeli (üks on punane, teine sinine).
Iga geeni jaoks on spetsiifiline kromosoom ja selle vastaspool, kuna see esineb kahes eksemplaris, mida nimetatakse alleelideks . Allel pärineb emalt ja elab ema kromosoomis; teine alleel pärineb isalt ja asub isa kromosoomis.
Geenidest pärinevad valgud, mis on meie kehas. DNA mutatsiooni ilmnemisel võib antud kromosoomi geen (tavaliselt alleel) olla defektne ja seega tekitada defektne valk või see võib puududa.
Mis on Prader-Willi sündroom?
Prader-Willi sündroom on haruldane geneetiline haigus, mida iseloomustab hulk füüsilisi, intellektuaalseid ja käitumuslikke puudujääke, mis sõltuvad 15. kromosoomi muutumisest.
Varajase lapsepõlve ajal ilmneb sündroomi ebatavalisest lihasnõrkusest ja arenguhäirest. Hiljem hakatakse lapsepõlves tekkima muid probleeme, nagu pidev isu, õpiraskused ja käitumishäired.
Prader-Willi sündroomiga inimesed on väga sageli ülekaalulisuse all kannatavad inimesed, kellest tekivad erinevad südameprobleemid. Need on peamised surma põhjused.
epidemioloogia
Prader-Willi sündroom on harvaesinev haigus: tegelikult on nakatunud lapse sünd sündinud keskmiselt iga 15 000-30 000 vastsündinu kohta.
See mõjutab mehi ja naisi võrdselt ning tal ei ole teatud tõugude suhtes eelsoodumust.
põhjused
Praderi-Willi sündroomi põhjuseks on geneetiline mutatsioon kromosoomil 15 . Täpset geeni ei ole veel selgitatud; kahtlused langevad rohkem kui midagi muud kromosomaalses piirkonnas, mis sisaldab rohkem geene.

Joonis: kromosoom 15 ja geenid, millel on kahtlus, et neil on Praderi-Willi sündroomi roll. Veebisaidilt: www.kreatech.com
CHROMOSOMA GENESID 15
Tavaliselt kasutavad meie keha rakud valkude loomiseks mõlemat alleeli. Teisisõnu tähendab see, et mõlemad kromosoomid, emad ja isad, on kasulikud ja annavad oma geneetilise panuse.
Kuid mõnedes konkreetsetes rakkudes toimib evolutsioonilise ja mittepatoloogilise probleemi tõttu ainult üks (isa või ema) alleel ja selle töö on enam kui rahuldav. Mitteaktiivset alleeli nimetatakse vaikiks, just sellepärast, et see eksisteerib, kuid see ei väljendu ise.
Meie aju rakud sisaldavad 15 kromosoomi, kuid mõnes piirkonnas ekspresseeritakse ainult ema geneetilist joont, teistes aga ainult isalikku. Hüpotalamuses, mis on Praderi-Willi sündroomi eest vastutav aju piirkond, ekspresseeritakse tavaliselt ainult isa kromosoomi geene.
CHROMOSOMA 15 JA PRADER-WILLI SYNDROME
Geneetilise testimise käigus leiti, et Prader-Willi sündroomiga patsientidel puudub normaalne isa kromosoom 15. See on kahjulik hüpotalamuses, kus ainus aktiivne kromosomaaljoon on isa.
Hüpotalamuse põhifunktsioonid:
- Söögiisu reguleerimine
- Une-ärkamise rütmide reguleerimine
- Emotsionaalsete seisundite väljendamine
- Kehatemperatuuri reguleerimine
- Hormooni tootmine
Aga mis käivitab isa kromosoomi 15 talitlushäire või puudumise? On vähemalt kolm võimalikku põhjust:
- Isapoolse kromosoomi 15 spetsiifilise piirkonna puudumine: tegelikult puudub kromosoomil oluline osa.
- Kaks ema 15 kromosoomi. See anomaalia tekib embrüo moodustumise ajal tekkinud vea tõttu.
- Mõne isa kromosoomi geeni 15 muutus.
GENEETILINE JA HEREDITAARNE? VÕI GENETIKA?
Geneetikud peavad Prader-Willi sündroomi geneetiliseks haiguseks, sest uuringute kohaselt selgus, et kolm ülalmainitud mutatsioonirežiimi ei ole päritud vanematelt (kellel on normaalne kromosomaalne varustus), vaid tekivad juhuslikult, vahetult enne ravi ( sporaadiline mutatsioon ).
Selle avalduse õigsust õõnestab aga mõnede vanemate paaride avastamine, kellel on rohkem kui üks laps, kes kannatavad Praderi-Willi sündroomi all. Nendel juhtudel on põhjust arvata, et haiguse alguses võib olla veel pärilik komponent, mida tuleb veel tõestada.
PRADER-WILLI SYNDROME JA ANGELMAN'i SYNDROME
Prader-Willi sündroom on mõnes mõttes Angelmani sündroomi vastand: viimasel juhul ei toimi ema kromosoom 15 korralikult.
Sümptomid ja tüsistused
Lisateabe saamiseks: sümptomid Prader-Willi sündroom
Prader-Willi sündroom ilmneb juba esimeste sümptomite ja sümptomitega juba esimesel lapsepõlves (esimene eluaasta); sel perioodil põhjustab see peamiselt lihaste toonuse (hüpotoonia) vähenemist ja arenguhäireid. Aja jooksul läbib haigus omamoodi evolutsiooni, mis rikastab sümptomaatilist pilti veelgi.
CHILDHOOD
Peamised märgid esimese eluaasta jooksul on:
- Lihas-hüpotoonia . See tähendab, et lihaste toon on võrreldes tavalise olukorraga vähenenud: see ilmneb tavaliselt pehmete ja mitte väga reaktsioonivõimeliste jäsemetega, aga ka ema piima raske imemisega.
- Arengu viivitus . Seda soodustab hüpotoonia tõttu tekkinud imemise raskus.
- Strabismus .
- Iseloomulikud näoomadused . Mandli silmad, pea kitsenemine templites, suu allapoole ja õhuke ülemine huul.
- Stimulatsioonile reageerimise osaline või täielik puudumine . Laps on väsinud ja teda ei ole lihtne üles äratada.
Vanusest kuni vanuseni? ADULT
Alates esimesest eluaastast on tekkinud palju probleeme, millel võib olla dramaatilisi tulemusi.
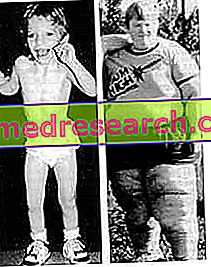
- Märkimisväärne isu ja rasvumine . Patsientidel on pidev soov toiduaine järele, mis viib neid palju sööma ja märkimisväärse kaalu. Kui nad ei leia midagi süüa, tulevad nad tarbima külmutatud toitu ja jäätmeid, teisisõnu midagi söödavat. Kõik see on tingitud hüpotalamuse muutunud funktsioonidest.
- Hüpogonadism . See tähendab, et suguelundid (munandid, inimestel ja munasarjadel naistel) toodavad vähe suguhormone (meessoost testosterooni ja naise östrogeeni). Patsient ei lõpeta puberteedi arengut ega ole tavaliselt viljakas. Esimene menstruatsioon naistel on hilinenud (kui mitte täielikult puudub); inimestel ei täheldata hääle muutust.
- Vähenenud majanduskasv ja areng . Püsiva lihaste hüpotoonia probleemile lisandub vähenenud staatiline areng isegi pärast puberteedi perioodi (kus tavaliselt noorukid tõuseb mitu sentimeetrit).
- Õpiraskused . Patsientide intellektuaalsed teaduskonnad on peaaegu alati vähenenud.
- Käitumisprobleemid . Eriti noorukieas on inimesed kangekaelsed, kapriissed ja kannatavad nn obsessiiv-kompulsiivse häire all.
- Mootori viivitus . Lapsed õpivad kõndima väga hilja.
- Keeleprobleemid . Tavaliselt hakkavad patsiendid rääkima märkimisväärse viivitusega ja nende keel jääb alati halvaks ja raskeks.
- Unehäired . Normaalset vaheldumist REM- ja NON-REM-unefaaside vahel ei järgita. Lisaks kannatavad patsiendid, kes magavad, hingamisraskuste (uneapnoe) all.
- Skolioos . Probleem on reserveeritud ainult mõnedele patsientidele.
KUIDAS VASTUTAB DOKTORILE
Imikul. Sümptomid, mis nõuavad Praderi-Willi sündroomi, on järgmised: arengu puudumine, lihas hüpotoonia, raskused rinnapiima imemiseks, näoomadused ja stiimulite puudumine.
Laps. Kaks põhilist vihjeid on: toidu pidev otsimine ja kiire kaalutõus.
TÜSISTUSED
Prader-Willi sündroomi peamised tüsistused on tingitud rasvumisest ja kõigist sellega seotud probleemidest, nagu diabeet, südamehaigus, hüpertensioon, hüperkolesteroleemia, ateroskleroos jne. Veelgi enam, pideva toitumise kontekstis on patsiendil lihtne lämbuda, sest söömiskord on tarbitud.
Teine väga oluliste tüsistuste seeria on seotud hüpogonadismiga : suguhormoonide puudumine põhjustab sageli steriilsust ja osteoporoosi .
diagnoos
Enne geneetiliste testide tegemist võib lihtsa füüsilise läbivaatuse ja mõnede vereanalüüside abil tuvastada Prader-Willi sündroomi õige diagnoosimise.
Kliinilised tunnused, mis tuleb leida füüsilise kontrolli käigus
imikul:
- Lihas-hüpotoonia
- Mandli silmad
- Templite vähenemine
Laps / nooruk:
- Talumatu isu
- ülekaalulisus
- Käitumisprobleemid
Geneetilised testid on kinnituseks ja aitavad selgitada haiguse põhjustanud mutatsiooni tüüpi.
ravi
Kahjuks, kuna see on geneetiline haigus, ei ole Prader-Willi sündroom ravitav.
Ainsad kohaldatavad raviprotseduurid on sümptomite (näiteks rasvumise) piiramine, mõõdukad mõned ebanormaalsed käitumised ja üldiselt parandada patsientide elatustaset.
Selleks, et see kõik õnnestuks, on soovitatav pöörduda arstide ja ekspertide meeskonda, kes on spetsialiseerunud erinevatele valdkondadele, alates endokrinoloogiast kuni dietoloogiani, alates füsioteraapiast kuni psühhoteraapiani.
Kõige tavalisemad ravimeetmed on loetletud allpool.
RAVIMINE LAPSES JA JÄRGMISES etappides
Varajase lapsepõlve ajal on imetamise raskuste ja arengu puudumise ületamiseks hea anda lastele kõrge kalorsusega toite.
Järgmistes etappides muutub olukord täielikult: manustatud toite tuleb hoolikalt kontrollida, pöörates maksimaalset tähelepanu kaloritele.
Kõige sobivam spetsialist nõu küsimiseks on dietoloog .
KASVU HORMONE
Kasvuhormooni ( GH ) eksogeensel manustamisel (st väljastpoolt) on kolm mõju:
- Kasvu soodustamine, mis muidu puuduks
- Parandage lihastoonust
- Vähendada keharasva taset
Ravi algab umbes 3-5 aastat.
Tänapäeval on laboris loodud hormonaalseid preparaate, mis on efektiivsed ja millel on väiksemad kõrvaltoimed.
Antud juhul on enim näidatud spetsialist endokrinoloog .
SOOVILISED HORMONID
Testosterooni eksogeenne manustamine meestele ja östrogeenidele naistele on oluline nende kahe hormooni vähendatud taseme taastamiseks. Lisaks viljakuse parandamisele on hormoonravil ka osteoporoosi vastane toime.
Ravi algab puberteedieas.
Lisateabe saamiseks: Prader-Willi sündroomi raviks kasutatavad ravimid »
Füsioterapia ja logopeedia
Prader-Willi sündroomiga patsiendid vajavad füüsilist ja keelelist rehabilitatsiooni . Esimene eesmärk on piirata lihaste hüpotooniat ja rasvumise mõju; teine õiguskaitsevahend kommunikatsioonipuudujääkidele, nii suulistele kui ka kirjalikele.
Eksperdid, keda pöörduda, on vastavalt füsioterapeudiks ja logopeediks.
PSÜHOTERAPIA JA TÖÖHÕIVEERAPIA
Psühhoteraapia on hädavajalik neile, kellel on obsessiiv-kompulsiivsed häired ja meeleolu üldiselt. Psühhiaatri või psühholoogi toetus võib oluliselt parandada käitumist.
Tööteraapia eesmärk on aga õpetada patsiendile, kuidas iseenda eest hoolitseda, kuidas riietuda jne, teisisõnu, kuidas teha peamisi igapäevaseid tegevusi.
Perekondade abi
Perekonnaliikmete lähedus on hädavajalik haige sugulase aitamiseks, eriti tema nooruse ajal. Nõuanded, mida tavaliselt antakse peredele, on patsiendi jälgimine kõigis oma tegevustes (eriti siis, kui ta on toidetud), küsida, milline on tema jaoks kõige sobivam käitumine, mitte teda välja jätta jne.
Prognoos ja ennetamine
Arvestades, et Praderi-Willi sündroom on ravimatu haigus, ei pruugi prognoos olla positiivne. Suurimat ohtu esindavad ülekaalulisus ja sellega seotud patoloogiad: surm on tavaliselt tingitud ühest neist.
Kättesaadavad ravid (tasakaalustatud toitumine, hormonaalne ravi, psühhoteraapia jne) parandavad elukvaliteeti isegi tundlikul viisil; siiski on ikka veel sümptomite kontrolli ja mitte midagi.
Sugulaste lähedus on oluline: nende toetus võib tegelikult patsientide eluiga pikendada.
ENNETAMINE
Kui haigus tekib embrüos geneetilise mutatsiooni tõttu, ei ole seda võimalik vältida.
Kui selle asemel on kaks vanemat juba sünnitanud Prader-Willi sündroomiga lapse, võivad nad enne teist rasedust teha spetsiifilisi geneetilisi teste, et teada saada, kas nad on haiguse kandjad või mitte.